Behandling av manifestationer vid myosit
Arbetsgrupp:
Ingrid Lundberg, Reumatologi, Karolinska Universitetssjukhuset och Karolinska Institutet, Stockholm
Tao Jin, Reumatologi, Sahlgrenska Universitetssjukhuset, och Sahlgrenska Akademin vid Göteborgs Universitet
Jehns Christian Martineus, Reumatologkliniken, Universitetssjukhuset, Lund
Danijel O´Rourke, Reumatologkliniken, Linköpings Universitetssjukhus, Region Östergötland
Elisabeth Skoglund, Reumatologkliniken, Akademiska sjukhuset och Uppsala Universitet, Uppsala
Bengt Wahlin, Norrlands Universitetssjukhus och Umeå Universitet, Umeå
Adjungerade till arbetsgruppen:
Helene Alexanderson, Karolinska Universitetssjukhuset och Karolinska Institutet, Stockholm
Olof Danielsson, Neurologiska kliniken, Universitetssjukhuset, Linköping
Ólöf Elíasdóttir, Neuromuskulärt centrum, Sahlgrenska
Sammanfattning
Idiopatisk inflammatorisk myopati, eller myosit, är en sällsynt reumatologisk, autoimmun sjukdom som kan påverka flera olika organsystem såsom muskler, hud, leder, lungor, hjärta och magtarmkanalen med åtföljande funktionsnedsättning och sänkt livskvalitet. Baserat på klinisk bild, serologi och muskelbiopsifynd kan myositsjukdomen hos vuxna indelas i flera olika subdiagnoser där prognos och behandlingssvar varierar; dermatomyosit, amyopatisk dermatomyosit, polymyosit, antisyntetassyndrom, immunmedierad nekrotiserande myopati, inklusionskroppsmyosit och overlap myosit. Då sjukdomsbilden ofta är komplex rekommenderas nära samarbete med vårdenheter som har team med erfarenhet av patienter med myosit och berörda organspecialister. Behandling av myosit är baserad på en kombination av läkemedelsbehandling och fysisk träning. Basen för läkemedelsbehandlingen vid myosit utgörs av kortison i höga doser över lång tid i kombination med immunmodulerande läkemedel. Här har vi indelat behandlingsrekommendationerna dels efter organmanifestationer och dels efter vissa subdiagnoser där speciell behandlingsstrategi är att rekommendera. Vi har även lagt in ett avsnitt om fysisk träning.
Innehåll
Behandling av manifestationer vid myosit
Bakgrund
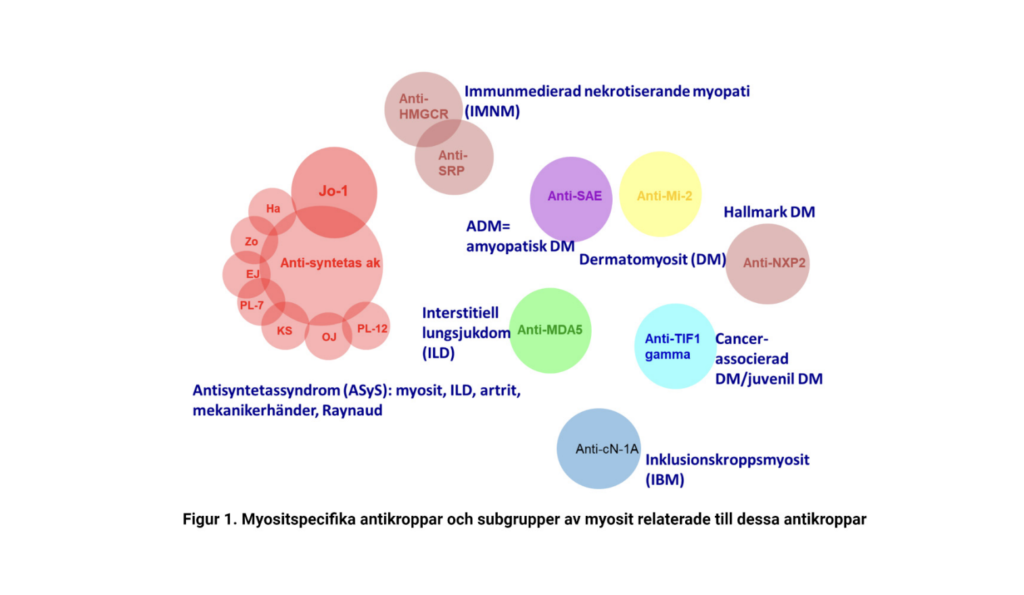
Idiopatisk inflammatorisk myopati (IIM) ofta kallad myosit, är en sällsynt inflammatorisk systemsjukdom med en incidens om ca 11 per miljon personår och en prevalens om ca 14 per 100 000 enligt svenska registerdata. Myosit är ett heterogent sjukdomstillstånd där kardinalsymtomet är muskelsvaghet som ses hos de flesta patienter. Extramuskulära symtom är vanliga och kan ibland dominera sjukdomsbilden, såsom hudutslag, interstitiell lungsjukdom, artrit, Raynauds fenomen och hjärtpåverkan. Sedan 50 år tillbaka har myositsjukdom hos vuxna indelats i dermatomyosit (DM), polymyosit (PM) och inklusionskroppsmyosit (IBM), en indelning baserad på skillnader i kliniska manifestationer och histopatologisk bild i muskelbiopsi. Autoantikroppar är vanligt förekommande vid myosit. Under de senaste decennierna har flera autoantikroppar identifierats som är specifika för myosit och sällan förekommer vid andra sjukdomstillstånd. Dessa autoantikroppar är ett starkt stöd för myositdiagnosen, dessutom är de associerade med specifika konstellationer av organmanifestationer och har gjort det möjligt att indela myosit i mer homogena subgrupper än de tidigare; därför har vi nu subgrupperna dermatomyosit, amyopatisk dermatomyosit, antisyntetassyndrom, immunmedierad nekrotiserande myopati (IMNM), inklusionskroppsmyosit, polymyosit och s.k. overlap myosit (OM) (Figur 1) (1). Med ökad kunskap om patofysiologi vid myosit är det möjligt att ytterligare subgrupper kommer att kunna identifieras.
Autoantikropparna kan indelas i myositspecifika och myositassocierade. De myositspecifika kan vidare indelas i antisyntetasantikroppar (anti-Jo1, anti-PL7, anti-PL12, anti-EJ, anti-OJ, anti-Zo, anti-HA och anti-KS), antikroppar vid dermatomyosit (anti-Mi-2, anti-SAE, anti-MDA5, anti-TIF1 gamma och anti-NXP2, där de senare två är associerade med cancer) antikroppar vid immunmedierad nekrotiserande myopati (anti-SRP och anti-HMGCR) och antikroppar associerade med inklusionskroppsmyosit, (anti-cN-1A) (Figur 1). En ny autoantikropp som ännu inte är tillgänglig i klinisk rutin är anti-FHL1 antikroppen. Myositassocierade autoantikroppar som också kan finnas vid andra reumatiska systemsjukdomar innefattar: anti-U1RNP, anti-Ro, anti-La, anti-Ku och anti-PM-Scl.
Den höga förekomsten av autoantikroppar tillsammans med att T lymfocyter ofta påvisas i muskelbiopsier och en mycket stark association till HLADRB1* allel ger stöd för att myosit är en autoimmun sjukdom. Behandlingen vid myosit är baserad på farmakoterapi, fysisk träning och annat stöd som kan erbjudas av det reumatologiska teamet. Myositsjukdomar är sällsynta och komplexa sjukdomar och har ibland livshotande manifestationer, därför bör de handläggas på eller i nära samverkan med universitetsklinik där tillgång finns till specialister inom lungmedicin, hudsjukdomar, kardiologi, neurologi och patologi samt det reumatologiska teamet: läkare, sjukgymnast, arbetsterapeut, kurator och sjuksköterska. Många patienter har sväljningsproblem och kan få bra stöd och hjälp av logoped.
Diagnosen myosit baseras på den sammantagna kliniska bilden med subakut insjuknande med objektiv proximal muskelsvaghet i övre och nedre extremiteter och nackflexorer, samt tecken på myopati i form av förhöjda muskelenzymer i serum (CK, LD, ASAT, ALAT eller aldolas), myopati/myosittecken på EMG, ödem på MRI-bild (T2-viktade bilder) samt tecken på inflammation samt regenererande och/eller degenererande fibrer i muskelbiopsi. Vid dermatomyosit kan olika hudutslag förekomma. De som anses mest specifika och utgör stöd för diagnosen dermatomyosit är Gottrons tecken och Gottrons papler, samt det heliotropa utslaget runt ögonen. Övriga hudutslag, dock inte specifika för myosit, är det s.k. V-tecknet, sjal-tecknet eller hölstertecknet och överväxt av nagelbanden. Typiska symtom och tecken för de övriga subgrupperna av myosit presenteras under respektive subgrupp.
Behandling vid myosit
De förslag på behandlingsrekommendationer som vi presenterar här är baserade dels på nyligen publicerade riktlinjer utgivna av British Society for Rheumatology (2), Norsk reumatologisk förenings guidelines, UpToDate och dels från PubMed. Det finns få kontrollerade studier varför rekommendationerna för behandling till stor del grundar sig på öppna studier eller retrospektiva studier och expertutlåtanden. Vidare är de flesta publicerade studier baserade på den äldre subgrupperingen av myosit: polymyosit, dermatomyosit och inklusionskroppsmyosit och än färre studier finns för de nya subgrupperna. Vårt dokument är inriktat på behandling av vuxna individer med myosit. Myosit kan debutera i barnaåren, varav många går i remission, men en del med juvenilt debuterande myosit utvecklar en kronisk form som fortsätter att ge symtom i vuxen ålder, antingen i form av fortsatta skov med aktiv inflammation eller i form av bestående skador t.ex. i form av muskelatrofi. Det finns inga studier att basera förslag till behandling av dessa personer, utan de får behandlas utifrån individuella bedömningar.
Vi ger först en översikt av farmakologisk behandling av muskelinflammationen vid myosit (baserad på studier med den äldre subgruppsindelningen), därefter följer behandlingsförslag för specifika manifestationer och kliniska subgrupper där det finns någon form av underlag för rekommendationer följt av råd om fysisk träning vid myosit.
Gemensamt för samtliga subdiagnoser av myosit är vikten av standardiserad utvärdering vid uppföljning. Samsjuklighet vid myosit, som kan förekomma vid de olika subgrupperna såsom ökad risk för hjärt-kärlsjukdom behandlas också i detta bakgrundsavsnitt, liksom korta rekommendationer för cancer-screening.
Utvärdering av behandling
Effekt av behandlingen bör utvärderas på ett standardiserat sätt och efter vilka organmanifestationer som dominerar sjukdomsbilden. Den internationella, multidisciplinära gruppen International Myositis Assessment and Clinical studies (IMACS) rekommenderar i kliniska studier uppföljning av sjukdomsaktivitet, organskada samt hälsorelaterad livskvalitet. Dessa mått är värdefulla att använda i klinisk praxis, särskilt kombinationsmåttet för sjukdomsaktivitet, vilket utgörs av ett sammantaget utfallsmått baserat på: läkarens övergripande bedömning på en visuell analogskala (VAS) 0-10 cm, patientens bedömning av sjukdomsaktivitet (i den svenska versionen övergripande hälsa) på en VAS-skala (0-10 cm), health assessment questionnaire (HAQ), manual muscle test (MMT) i 8 muskelgrupper, max score 80, (mmt8_grading_and_testing_procedures_for_the_abbreviated_8_muscle_groups (nih.gov)) muskelenzymvärden i serum (2 av följande: CK. ASAT, ALAT, LD eller aldolas) samt ett extramuskulärt score på en VAS skala 0-10 cm, alternativt Myositis disease activity assessment tool MDAAT= som är baserat på 6 domäner (3). Dessa variabler utgör grunden för det i consensus föreslagna Total Improvement Score (TIS), som nu ofta utgör Primary endpoint i kliniska läkemedelsstudier (4).
Utöver detta kan Functional Index 2 (FI-2) eller Functional Index 3 (FI-3) användas, som mäter funktionell dynamisk uthållighet och utgör en kompletterande undersökning av muskelfunktion då MMT har takeffekt, det vill säga att trots uppmätt maxvärde (=80) kan man ha en funktionsnedsättning (5).
Functional Index-2: Scoring Sheet and Instructions (nih.gov)
Functional Index-2: Training Guide (nih.gov)
Instruktionsfilm: Functional Index 2 Myositis – YouTube
Vidare kan greppstyrka utvärderas med Grippit eller Jamar och handens finmotorik kan mätas med perdue pegboard. Självrapporterad livskvalitet mäts med RAND-36. Samtliga dessa mått finns i besöksmodulen i SRQs myositmodul.
UTVÄRDERING av terapieffekt på sjukdomsaktivitet bör göras efter 3 månader. Om då ingen förbättring har uppnåtts bör i första hand diagnosen ifrågasättas, i nästa steg bör terapibyte övervägas. Det är vid uppföljning viktigt att försöka skilja på orsaken till kvarstående muskelsvaghet; kvarvarande aktiv sjukdom eller symtom till följd av skada orsakade av tidigare inflammation, då behandlingen skiljer sig helt.
Hjärt-kärlmanifestationer
Patienter med myosit kan drabbas av hjärtengagemang i form av myokardit eller perikardit. Tillståndet är ofta asymtomatiskt. Varken EKG eller UCG är särskilt känsliga för att upptäcka hjärtengagemang, särskilt inte vid lindrig sjukdom. Cardiac troponin I (cTnI), men inte troponin T (cTnT), är en känslig och specifik biomarkör för tidig upptäckt av myokardit vid myosit (2, 6). Kardiell magnetresonanstomografi (CMR) utgör ”gold standard” för att verifiera myokardit eller annan hjärtinflammation vid misstanke om hjärtengagemang (7, 8).
Prognosen vid hjärtengagemang är ännu ofullständigt studerad. Behandling vid hjärtengagemang är inte heller fastställd, men kortikosteroider och DMARDs som används vid myosit kan troligen även dämpa den inflammatoriska processen i hjärtat, vilket avspeglas i sjunkande cTnI-nivåer och regress av myokardit (opublicerade data, Göteborg). Vid uttalat eller progredierande hjärtengagemang bör handläggningen ske i nära samarbete med kardiolog.
Patienter med myosit har en ökad risk för kardiovaskulär sjukdom. Sannolikt beror denna ökade risk både på den kroniska inflammationssjukdomen som i sig kan drabba hjärtmuskeln och på långvarig kortisonbehandling. Traditionella kardiovaskulära riskfaktorer såsom hypertoni, diabetes, lipidrubbningar, och fetma är vanligt förekommande hos patienter med IIM (9-12). En retrospektiv populationsbaserad kohortstudie har visat att IIM är associerat med en ökad risk för kardiovaskulära händelser under de första fem åren efter diagnos. Risken är lika hög som vid RA (13). Vi rekommenderar att patienter med IIM bör screenas och behandlas för att förebygga aterosklerotisk hjärt-kärlsjukdom enligt SRFs rekommendation för kardiovaskulär primärprevention vid inflammatorisk reumatisk sjukdom. Patienter med myosit har också en ökad risk för venös tromboembolism (VTE) särskilt de som har en cancerassocierad myosit och särskilt under första året efter myositdiagnos. Denna risk bör finnas i åtanke vad gäller klinisk handläggning av dessa patienter.
Cancerscreening
Patienter med myosit, särskilt subgruppen dermatomyosit har en ökad risk för cancer. Detta gäller särskilt vuxna personer med dermatomyosit, i ålder över 40 år, och särskilt de personer som har anti-TIF1-gamma eller anti-NXP2 autoantikroppar. Vid förekomst av anti-TIF1-gamma antikroppar är cancerrisken särskilt hög i tidsintervallet 3 år före och 3 efter dermatomyositdiagnos. Internationella guidelines för cancerscreening för patienter med myosit har nyligen publicerats (14). Patienter med inklusionskroppsmyosit har inte befunnits ha en ökad risk för cancer. Alla andra vuxna individer med myositdiagnos bör genomgå en basal cancerscreening vid myositdiagnos, riktad efter kliniska manifestationer och avvikelser i provsvar samt enligt nationella screeningriktlinjer för cancer. Patienter med antisyntetassyndrom eller patienter med myosit i kombination en annan reumatisk sjukdom (overlapmyosit) anses ha låg cancerrisk och för dessa gäller basal cancerscreening. För övriga personer med myosit, rekommenderas därutöver screening med CT hals, thorax, buk, och bäcken, F-Hb och serumtest av CA-125. För kvinnor tillkommer mammografi och gynekologundersökning med transvaginalt ultraljud och smear test och för män PSA-test. För individer med hög risk för cancer såsom de som är positiva för anti-TIF-gamma eller anti-NXP2 autoantikroppar eller har andra riskfaktorer för cancer bör basal cancerscreening upprepas efter 1,2 och 3 år från myositdiagnos. Riskstratifiering och screeningfrekvens: se länk: Risk stratification and frequency of screening for IIM-related cancer.
Vid fastställd cancerdiagnos bör behandling av patientens myosit ske i samverkan med behandlande onkolog. Ofta förbättras myositen om cancersjukdomen behandlas framgångsrikt.
Förkortningar
ASyS, antisyntetassyndrom
CDASI, The Cutaneous Dermatomyositis Disease Area and Severity Index
DLCO diffusing capacity of the Lungs for Carbon Monoxide, Diffusionskapacitet
DM, dermatomyosit
DMARD, disease modifying anti-rheumatic drug
ECMO, extracorporeal membranoxygenering
EMG, elektromyografi
FI, Functional Index
FVC, Forced vital capacity
IBM, inklusionskroppsmyosit
IIM, Idiopatisk inflammatorisk myopati
IMACS, International Myositis Assessment and Clinical studies
IMNM, immunmedierad nekrotiserande myopati
MDAAT, Myositis disease activity assessment tool
MITAX, Myositis intention to treat activity index
MMT, Manual muscle test
MRI, magnet resonance imaging
mTOR, mammalian target of rapamycin
PM, polymyosit
CAR-T, Chimeric Antigen Receptor-T cell
Behandling av muskelinflammationen vid myosit (dermatomyosit, antisyntetassyndrom och polymyosit)
Bakgrund
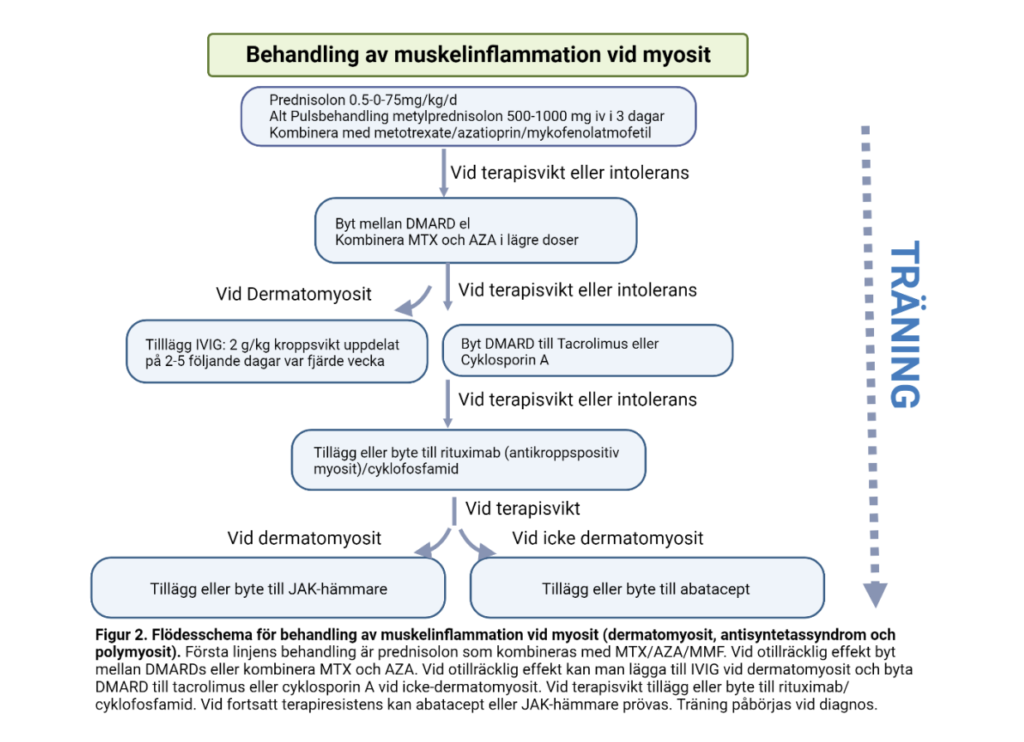
De flesta rekommendationer för behandling av muskelinflammationen vid myosit föreslår att immundämpande behandling påbörjas så snart som möjligt efter att diagnos myosit har fastställts. Här är subgruppen inklusionskroppsmyosit (IBM) ett undantag, se separat text om IBM. Basen i farmakologisk behandling av myosit utgörs av induktionsbehandling med glukokortikoider i kombination med ett immundämpande läkemedel (disease modifying anti-rheumatic drug, DMARD) för att minska kortisonbehov och för att optimera den immundämpande effekten. Oftast föreslås metotrexate som förstahandspreparat. Alternativt kan azathioprin eller mykofenolatmofetil användas. I nästa steg kan dessa preparat bytas mot varandra. Som andrahandsval, kan två förstahandsmedel kombineras (metotrexate och azathioprin) eller ersättas med cyklosporin eller takrolimus. I tredje hand föreslås tillägg med rituximab eller cyklofosfamid eller vid subgruppen dermatomyosit: högdos intravenöst immunglobulin där en nyligen publicerad randomiserad, kontrollerad studie visade signifikant effekt med det primära utfallsmåttet ”total improvement score” hos patienter med dermatomyosit (15). Vid fortsatt terapiresistens eller intolerans kan abatacept eller annat preparat som ingår i pågående läkemedelsstudier prövas (Fig 2). För vissa subgrupper finns andra rekommendationer baserade på erfarenhet, v.g. se nedan.
Det finns tydlig evidens för att farmakologisk behandling bör kombineras med fysisk träning såsom beskrivs i ett eget avsnitt.
Farmakologisk behandling av muskelinflammation
KORTISON utgör grunden i behandlingen. Behandling av muskelinflammationen föreslås påbörjas med prednisolon 0,5-0,75 mg/kg kroppsvikt per dag, ofta ca 40- 60 mg prednisolon/dag i kombination med ett immunmodulerande preparat. Ett alternativ till induktionsbehandling är intravenös pulsbehandling med metylprednisolon i 3 dagar, följt av peroralt kortison i lägre startdos. Nedtrappning av kortisondosen kan påbörjas när patienten har börjat förbättras i muskelstyrka/muskulär uthållighet. Ofta behövs 4 veckors behandling, ibland 6 veckor med högdos för att se effekt på muskelfunktion, men i fall med snabb klinisk förbättring kan kortisonnedtrappning påbörjas efter ca 2 veckor. Vidare evidens saknas för hur kortisonnedtrappningen kan ske. Nedtrappningen bör dock genomföras baserat på den sammantagna kliniska bilden där muskelfunktion mätt med MMT-8 eller FI-2 eller FI-3 är det viktigaste måttet. Observera att serumnivåer av CK, hos vissa individer kan sjunka snabbt vid insatt kortisonbehandling utan samtidig förbättring av muskelfunktion. CK-nivåer ska därför inte styra den immundämpande behandlingen. Hur länge den immundämpande behandlingen behöver pågå varierar mellan individer. Utsättning av immundämpande läkemedel bör prövas då sjukdomen varit i remission i minst 1 år med försiktig dosminskning och tät uppföljning av kliniska manifestationer. De flesta patienter behöver dock mångårig eller livslång behandling med immundämpande läkemedel, ibland i låga doser. Behandling av glukokortikoidinducerad sekundär osteoporos bör följa Läkemedelsverkets riktlinjer för läkemedelsbehandling vid osteoporos (https://www.lakemedelsverket.se/4ab04f/globalassets/dokument/behandling-och-forskrivning/behandlingsrekommendationer/behandlingsrekommendation/behandlingsrekommendation-osteoporos.pdf), i syfte att förebygga benskörhetsfrakturer.
Första linjens behandling
METOTREXATE är det immundämpande läkemedel (csDMARD) som oftast används som första linjens behandling i kombination med prednisolon. Metotrexate ges peroralt eller subkutant upp till 25 mg/vecka. Tillsammans med metotrexate rekommenderas tillägg med folsyra såsom vid reumatoid artrit. Vid utebliven effekt av metotrexate kan detta bytas mot något av nedanstående csDMARD, alternativt kombineras med azathioprin, men då i lägre dos för båda preparaten för att minska risk för biverkningar.
AZATHIOPRIN kan ges i kombination med prednisolon, vanligtvis i dosen 2 mg/kg kroppsvikt.
MYKOFENOLATMOFETIL kan ges i kombination med prednisolon, vanligtvis i dosen 2000 mg /dag men dosen kan ökas upp till 3000 mg/dag, ges i delad dos.
Andra linjens behandling
TACROLIMUS eller CYKLOSPORIN A, är båda preparat som bland annat hämmar T lymfocyter. Båda bör ges 2 gånger per dag. Takrolimus bör doseras till ett dalvärde i koncentration på 5-10 ng/ml, start dos 1-3 mg/dygn. Cyklosporin har använts i doser på 2-5 mg/kg kroppsvikt. Cyklosporin kan också doseras till ett dalvärde på 100-150 ng/ml.
INTRAVENÖST IMMUNGLOBULIN i högdos, som tillägg till DMARD kan övervägas till patienter med terapiresistent dermatomyosit. Det finns evidens för att en dos av 2 g/kg kroppsvikt var fjärde vecka under 16 veckor ger åtminstone minimal förbättring av Total Improvement Score (TIS) som återspeglar förändringen över tid i dermatomyositaktivitet. Detta baseras på resultat från en randomiserad och kontrollerad studie (15).
Tredje linjens behandling
RITUXIMAB kan övervägas till patienter med svår och terapiresistent myosit, särskilt subgrupper med autoantikroppar och till patienter med låg grad av organskada, som tillägg till csDMARD eller istället för csDMARD. Dosen som användes i ”Rituximab vid Myositstudien” var 1000 mg i.v. med 2 veckors mellanrum som induktionsterapi följt av 1000 mg i.v. efter 6 månader (16).
CYCLOFOSFAMID givet intravenöst kan övervägas vid svår och terapiresistent myosit. Olika protokoll har använts såsom Eurolupusprotokollet eller enligt vaskulitbehandling.
Fjärde linjens behandling
ABATACEPT, kan övervägas vid polymyosit, immunmedierad nekrotiserande myopati och antisyntetassyndrom (Abstrakt ACR 2022).
JAK-HÄMMARE (tofazitinib) vid dermatomyosit
En nyligen genomförd systematisk litteraturöversikt antyder att JAK-hämmare är effektiva för både hudmanifestationer och muskelinflammation vid dermatomyosit (17). Tofacitinib framstår som särskilt effektivt för hudmanifestationer vid dermatomyosit och amyopatisk dermatomyosit, enligt en retrospektiv studie. Det bör dock noteras att det endast observerades minimal nytta för muskelstyrka i denna studie (18).
Enstaka fallrapporter har visat mycket bra effekt vid svårbehandlat antisyntetassyndrom på lungmanifestationer och på muskelstyrka med CD19 CAR-T cell behandling (19).
Behandling av interstitiell lungsjukdom vid myosit
Bakgrund
Interstitiell lungsjukdom (ILD) är en mycket allvarlig manifestation av myositsjukdomen med hög morbiditet och mortalitet. Förekomsten av ILD har för patienter med dermatomyosit och polymyosit som helhet uppskattats till ungefär 40 % (20), men varierar bland annat beroende på antikroppsprofil. Den är betydligt högre vid exempelvis antisyntetassyndromet där 80 % av patienterna med Jo1-antikroppar utvecklar ILD (21). Förloppet varierar från asymptomatisk ILD till snabbt progredierande sjukdom, där MDA-5-positiv dermatomyosit utmärker sig med hög risk för snabb progress särskilt hos individer från ostasiatisk population såsom Japan och Kina.
Givet hur vanlig och allvarlig ILD är, bör patienter med myosit utredas för misstänkt ILD vid diagnos och vid nytillkomna luftvägssymptom eller avvikande fynd vid lungauskultation. Den radiologiska metod som i första hand bör användas är datortomografi med tunna snitt (HRCT), eftersom den är betydligt känsligare än konventionell röntgen. Lungfunktionsundersökning med mätning av diffusionskapaciteten (DLCO) bör också göras vid nydiagnosticerad myosit. DLCO bör vara korrigerat för Hb. Det är värt att notera att DLCO är ett känsligare mått på ILD än vitalkapaciteten, och inte heller påverkas av funktionen i andningsmuskulaturen i samma utsträckning som denna. Man bör också beakta att det föreligger en stor variation i både vitalkapacitet och DLCO hos friska individer, varför en individ med diskret ILD kan ha normalfynd vid lungfunktionsundersökning. För att följa förloppet av ILD är dock lungfunktionsundersökning med DLCO en utmärkt metod. Det finns inte evidens för att rekommendera i vilken mån screening för ILD ska upprepas hos patienter med myosit, men HRCT och lungfunktionsundersökning ska göras vid exempelvis nytillkomna patologiska fynd vid lungauskultation, sjunkande fysisk arbetsförmåga eller nytillkomna luftvägssymptom. Förekomst av antisyntetasantikroppar, anti-MDA5, anti-KU, anti-PM/SCL eller anti-Ro52 autoantikroppar medför en ökad risk för ILD, varför upprepad screening med HRCT och lungfunktionsundersökning bör övervägas för patienter med dessa antikroppar(22). Infektioner, inklusive opportunister, ska också has i åtanke vid sjukdom i lungor och luftvägar hos patienter med myosit som behandlas med kortison och andra immunmodulerande läkemedel. Profylax mot Pneumocystis jirovecii bör ges, särskilt vid höga kortisondoser och vid kombination av flera immunmodulerande preparat.
ILD klassificeras ofta baserat på utseendet vid HRCT. Vid myosit är non-specific interstitial pneumonia (NSIP) det vanligaste mönstret, precis som det är vid flertalet andra systemsjukdomar. Även andra mönster förekommer dock, inte minst usual interstitial pneumonia (UIP) som är associerat med sämre prognos i ILD. Även organizing pneumonia (OP) förekommer vid myosit (23).
Farmakologisk behandling av ILD
Det finns mycket få kontrollerade studier avseende behandling av ILD vid myosit, utan de publikationer som finns är i stor utsträckning fallrapporter och fallserier. Detta gör att rekommendationer om behandling är baserade på ett magert vetenskapligt underlag, men de sammanfattar ändå den kunskap som är tillgänglig. Under 2025 har guidelines för handläggning av ILD vid systemsjukdomar, inkluderande myosit, publicerats (24). Det är också värt att ha i åtanke att ILD vid myosit inte är någon enhetlig sjukdom, utan utgörs av olika lungmanifestationer med varierande grad av kliniska symtom, från asymptomatisk till snabbt progredierande och med olika grad av inflammation och fibros, vilket medför att de observationer som rekommendationerna baseras på är heterogena. Det finns dock övertygande stöd för att immunmodulerande behandling är till gagn för patienter med myositassocierad ILD, både i termer av minskad lungsjukdom, förbättrad lungfunktion men också genom minskat kortisonbehov. Viktigt är att dessa patienter handläggs i samråd med lungläkare och gärna också med radiolog med speciell expertis inom lungsjukdomar. Diskussion inom interdisciplinära ronder rekommenderas särskilt vid komplicerade fall. Utvärdering av behandlingseffekt bör göras avseende klinik men också med lungfunktionstest mätt med FVC och DLCO efter 6 veckor och 3 månader, eller oftare efter individuell bedömning. Vid progress under pågående behandling bör ny HRCT övervägas för att bedöma orsak till försämringen.
Nedan följer beskrivning av de preparat som är aktuella (Figur 3).
Immunmodulerande behandling
Första linjens behandlingsval vid ILD
ILD vid myosit bör behandlas med kortison och ytterligare immunmodulerande behandling. Det finns inte stöd för att rekommendera något specifikt preparat, utan valet måste göras av behandlande läkare beroende på hur allvarlig lungsjukdomen är, samt övriga organmanifestationer och sjukdomar.
KORTISON ska insättas omedelbart vid behandling av myositassocierad ILD. Vanligen används prednisolon i doser på 0,5-1 mg/kg kroppsvikt, som sedan nedtrappas efter kliniskt svar (25, 26). Vid akuta tillstånd bör intravenös behandling med metylprednisolon 500-1000 mg användas. Kortison i monoterapi kan inte rekommenderas, utan tillägg av ytterligare immunmodulerande behandling ska också insättas.
AZATHIOPRIN är ett välbeprövat preparat som använts i stor utsträckning vid systemsjukdomar, inklusive myositassocierad ILD. Det finns också visst stöd för att det är effektivt vid detta tillstånd (27-29). Rekommenderad dos är 2 mg/kg kroppsvikt (26).
MYKOFENOLATMOFETIL (MMF) är också relativt vanligt vid myositassocierad ILD, och även här finns visst stöd för att behandlingen har effekt (28). MMF har inte bättre effekt än azathioprin i de mycket små och heterogena material där dessa är jämförda, men hade lägre frekvens av biverkningar än azathioprin (27, 28). Rekommenderad dos av MMF är minst 2000 mg/dygn, men upp till 3000 mg/dygn om det tolereras (26).
CALCINEURINHÄMMARE är de mest studerade preparaten. De aktuella preparaten är takrolimus och cyklosporin, som båda har samma verkningsmekanism. Det finns ett relativt stort antal studier, som visar att både cyklosporin och takrolimus har god effekt på myositassocierad ILD (30-32). Takrolimus och cyklosporin är jämförda sinsemellan i en open-label-studie, som visade en icke signifikant tendens till bättre effekt av takrolimus (25). Takrolimus kan startas i dosen 1-3 mg/dag och sedan doseras till ett dalvärde i koncentration på 5-10 ng/ml, medan cyklosporin har använts i doser på 2-5 mg/kg kroppsvikt (26). Cyklosporin kan också doseras till ett dalvärde på 100-150 ng/ml (25). Det finns stöd för att takrolimus har god effekt också när det adderas till pågående behandling med kortison och annat preparat, vanligen MMF eller azathioprin (29, 31).
METOTREXATE är ett förstahandspreparat vid myosit, men väldigt lite är beskrivet av dess effekter på ILD. En deskriptiv studie som omfattade 17 patienter behandlade med metotrexate antyder dock att metotrexate är effektivt mot ILD (29).
Andra linjens behandlingsval vid behandlingsrefraktär ILD vid myosit
I en nyligen publicerad randomiserad, kontrollerad fas 2b-studie där cyklofosfamid jämfördes med rituximab för behandling av ILD vid inflammatorisk systemsjukdom, ingick 44 patienter med myosit och ILD. Förbättring avseende lungfunktion mätt som FVC uppnåddes med båda preparaten utan signifikant skillnad, men det var färre biverkningar i rituximabgruppen (33). Inte heller en meta-analys där rutiximab och cyklofosfamid jämfördes kunde visa någon signifikant skillnad i effekt (34). Det har också publicerats en liten RCT där tillägg av abatacept inte visade någon signifikant positiv effekt på ILD hos patienter med antisyntetassyndrom (35).
CYKLOFOSFAMID har effekt på ILD vid myosit (28, 30, 36), men med välkända biverkningar och risker. I det lilla antal jämförande studier som finns, har det ej haft bättre effekt än MMF, azathioprin (28), cyklosporin (30), eller rituximab (36).
RITUXIMAB har visat god effekt på ILD vid myosit, men flertalet studier är gjorda på antisyntetassyndromet (36, 37). I en studie hade rituximab som induktions- och underhållsbehandling bättre effekt än induktion med cyklofosfamid följt av annan immunmodulerare (36).
Rituximab är också ett värdefullt tillägg till annan behandling, när så behövs. Den vid övriga reumatiska sjukdomar gängse initiala doseringen på två doser á 1000 mg med två veckors mellanrum därefter 500-1000 mg med 6 månaders mellanrum kan rekommenderas vid ILD vid myosit.
INTRAVENÖST IMMUNOGLOBULIN (IVIG) är icke tillfredsställande studerat vid myositassocierad ILD. Den största studien som publicerats omfattar 17 patienter och visar att IVIG har effekt som tillägg till pågående kombination av kortison och annan immunmodulerande behandling (27). IVIG kan därmed vara aktuellt vid svår ILD. En särskild indikation för IVIG kan vara ökad infektionsbenägenhet vid uttalad immunsuppression hos dessa patienter.
Tredje linjens behandlingsval vid behandlingsrefraktär ILD vid myosit
PLASMAFERES är inte studerat vid annan myositassocierad ILD än MDA-5-positiv dermatomyosit (se nedan under snabbt progredierande ILD).
FIBROSHÄMMARNA NINTEDANIB och PIRFENIDON kan ha effekt på progressiv fibrotiserande ILD. Behandling med fibroshämmare bör endast ske i samråd med lungläkare.
Snabbt progredierande ILD och MDA-5-positiv dermatomyosit
ILD vid MDA-5-positiv dermatomyosit är ofta ett mycket allvarligt tillstånd med snabbt progredierande ILD och en hög mortalitet. Patienter med snabbt progredierande lungfunktionsnedsättning bör handläggas i nära samverkan med lungläkare och diskussion om möjlighet till lungtransplantation bör initieras tidigt vid snabb försämring av lungfunktion. ECMO kan vara livräddande både i väntan på effekt av immunsuppression eller på lungtransplantation.
Första linjens behandlingsval vid snabbt progredierande ILD
Vid detta tillstånd rekommenderas snabbt insatt behandling med kortison (Figur 4). Visst stöd finns för calcineurinhämmare (i första hand takrolimus, men också cyklosporin) som induktionsbehandling. Tidigare studier har talat för att tidigt insatt kombination med calcineurinhämmare och cyklofosfamid, skulle vara mer effektiv än upptrappande terapi, men detta har inte bekräftats i en senare studie (38, 39). Alternativt rekommenderas som induktionsbehandling kombination av kortison med rituximab, eventuellt i kombination med MMF (2, 40). Det finns också visst stöd för att tillägg med plasmaferes är gynnsamt vid MDA-5-positiv ILD (41, 42). Enligt framför allt kinesiska och japanska studier kan tofacitinib ha effekt vid MDA-5-positiv ILD och det finns även fallserier som visar gynnsam effekt av baricitinib (39, 43, 44).
Andra linjens behandlingsval vid snabbt progredierande ILD
Vid bristande behandlingssvar rekommenderas byte av cyklofosfamid mot rituximab eller eventuellt en kombination av dessa två preparat, såsom redovisas i flödesschemat.
Behandling av DYSFAGI – Sväljningssvårigheter
Bakgrund
Att svälja är en komplex neuromuskulär funktion som innefattar orofarynx, larynx och esofagus. Den övre delen består av tvärstrimmig muskulatur medan den mellersta utgörs av glatt muskel. Eftersom idiopatisk inflammatorisk myopati (IIM) eller myosit, påverkar tvärstrimmig muskulatur är det inte så märkligt att en påverkan på sväljningsförmågan kan uppstå. Dysfagi beräknas förekomma hos 33 % av myositpatienter och hos upp till 77 % hos patienter med inklusionskroppsmyosit (IBM) (45). Det märkliga vid IBM är att cricofaryngeusmuskeln hypertrofierar till skillnad från andra involverade muskelgrupper vilket kan kräva operativa åtgärder. Dessutom är sväljningsproblem underrapporterade hos IBM patienter och uppträder oftast i det senare skedet av sjukdomen. Vid cancerassocierad myosit och anti-NPX2-positivitet är prevalensen av dysfagi 62 %.
Sväljningsbesvär kan förutom påverkan på livskvaliteten även leda till viktnedgång, dehydrering, aspirationspneumoni och mortalitet. Försämring av livskvaliteten kan yttra sig genom social isolering, undvikande att äta tillsammans med andra och rädsla för att svälja fel. Dessa patienter bör handläggas i samråd med öron-näsa-hals specialister och logopeder.
Farmakologisk behandling
Initial farmakologisk behandling kan innefatta högdos steroider framför allt i form av intravenöst metylprednisolon givet som pulsbehandling med eller utan kombination med metotrexate eller azathioprin (Figur 5).
Det finns några studier som visar att högdos IVIG är ett alternativ vid behandling av dysfagi hos patienter med IBM (46-49) men även hos patienter med dermatomyosit (DM) och en fallstudie har visat att subkutant immunglobulin (IG) kan vara ett alternativ (50). Det finns också fallrapporter om effekt av cyklofosfamid vid DM med dysfagi (51).
Kompletterande behandling
Förutom immunosuppressiv behandling kan andra alternativ vara esofagusdilatation eller vidgning av cricofaryngeusmuskeln med s.k. ballongdilatation. Även myotomi av cricofaryngeusmuskeln har utförts men görs numera sällan. Andra behandlingsalternativ är botulinuminjektion i cricofaryngeus eller elektrisk stimulering (transkutan nervstimulering av orofaryngeala muskler s.k. vital stim). Vid uttalade svårigheter att svälja får man ta ställning till nasogastrisk sond eller PEG. Logopeder kan ge viktiga tips hur patienter kan träna svalgmuskler samt också vad de ska tänka på när de ska svälja mat och dryck. Även kontakt med dietist rekommenderas.
Behandling av hudmanifestationer
Bakgrund
Dermatomyosit (DM) karaktäriseras utöver av progressiv, symmetrisk, proximal muskelsvaghet av typiska hudmanifestationer. De flesta patienter har samtidiga hud- och muskelsymptom. Hudsymptom kan dock föregå debut av myosit med upp till flera månader och i vissa fall kan hudmanifestationer utvecklas helt i frånvaro av muskelsjukdom. Denna subgrupp kallas amyopatisk DM eller vid vissa tecken på myopati tex förhöjt CK utan muskelsvaghet för hypomyopatisk DM. Hudmanifestationerna kan ibland kvarstå trots framgångsrik behandling av myosit. Gottrons papler och heliotropt exantem är patognomona hudmanifestationer hos patienter med DM. Dessutom får patienter ofta intensiv klåda med sammanflytande, kraftiga erytem i hårbotten, ansikte, övre bålen och övre extremiteter.
Förbättring av hudmanifestationer kan inträffa under behandling av andra manifestationer (myosit eller lungengagemang) av DM. Om det finns en ihållande aktivitet av kutan DM trots adekvat kontroll av extrakutan DM, så kan systemisk behandling specifikt för hudsymptom läggas till. Data om behandling för hudmanifestationer är begränsade och utgörs främst av fallrapporter och retrospektiva studier.
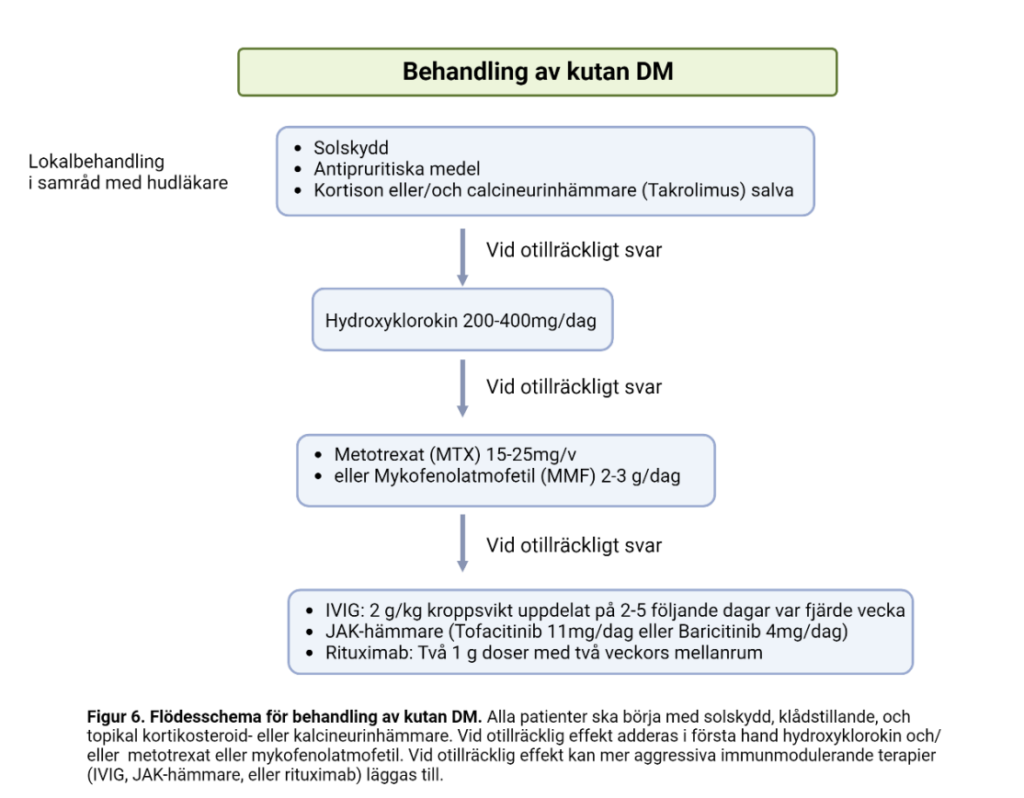
Initial behandling av hudmanifestationer vid DM inkluderar solskydd, klådstillande, och topikal kortikosteroid eller kalcineurinhämmare. Detta bör handläggas i samråd med hudläkare. Hudmanifestationer är ofta resistenta mot enbart solskydd och topikal behandling och kräver då DMARD-behandling så som hydroxyklorokin, metotrexate, eller mykofenolatmofetil. Patienter som inte svarar på dessa behandlingar kan behöva mer aggressiva immunmodulerande terapier (IVIG och rituximab) (Figur 6). En del patienter med DM utvecklar en svårbehandlad calcinos i hud eller subcutan vävnad. Calcinos ses främst hos individer med debut av DM i barnåren men förekommer ibland vid DM som debuterar hos vuxna, särskilt yngre vuxna med långvarig, svårbehandlad hudinflammation. Denna calcinos är ofta terapiresistent.
Första linjens behandlingsval vid hudutslag
Solskydd: Exponering för UV-ljus kan förvärra kutan DM (52, 53). Dagligt solskydd året runt med solskyddsmedel med en solskyddsfaktor (SPF) på minst 30 rekommenderas (54).
Antipruritiska medel: Klåda kan vara besvärlig hos patienter med DM och kan ha betydande negativa effekter på livskvalitet. Klåda bör behandlas med topikala eller orala antipruritiska medel:
- Mjukgörande och klådstillande krämer eller kutan emulsion, såsom Canoderm, Kräm Karbamid i Essex APL, Kräm Karbasal och Lotion Propyless.
- Orala antipruritiska medel – antihistaminer (Tavegyl, Atarax, och Desloratadin).
Lokal behandling
Topikala kortikosteroider kan ges till patienter med hudbesvär i hårbotten, bålen och extremiteter (55). Topikala kalcineurinhämmare (Takrolimus 0,1% salva) kan användas till patienter som inte förbättras med topikala kortikosteroider eller i hudområden som är benägna att utveckla kortisoninducerad kutan atrofi, såsom ansiktet (56, 57).
Andra linjens behandlingsval vid hudutslag
DMARD
Hydroxiklorokin (200mg-400mg/dag) är förstahandsterapi för mild kutan DM (58, 59). Rökning kan minska effekten av antimalarialäkemedel, och därför bör rökavvänjning uppmuntras (60). En analys av 115 patienter med amyopatisk eller hypomyopatisk DM tyder dock på att hydroxiklorokin har begränsad effektivitet för många patienter med kutan DM då endast 11% av patienter uppnådde kontroll av hudmanifestationer (58).
Metotrexate (MTX): Effekten av MTX för hudmanifestationer av DM har inte studerats i randomiserade studier, och evidensen är begränsad till retrospektiva analyser (61, 62). I en studie av 13 patienter med kutan DM som behandlades med MTX (2,5 till 30 mg/vecka), uppnådde 8 patienter remission i hudsymptom (62). I en annan studie fick 8 av 11 patienter med kutan DM som behandlats med MTX en markant förbättring i Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) score med minskat lymfocytär infiltration i hudbiopsi (61).
Mykofenolatmofetil (MMF) (2-3g/dag) som används för behandling av myosit vid DM är också fördelaktigt för hudsymptom. I en prospektiv studie av 74 patienter med måttlig till svår kutan DM som behandlades med olika DMARDs fann man ett samband mellan MMF behandling och klinisk remission (63). Dessutom visade denna studie att majoriteten av patienterna som uppnådde klinisk remission med MMF behandlades med en högre dos (1.5gx2). I en retrospektiv studie av 12 patienter med kutan DM som var refraktär mot andra DMARDs (hydroxiklorokin, MTX, och azatioprin), gav MMF klinisk förbättring hos 10 patienter (64).
Intravenöst immunglobulin – IVIG (2g/kg kroppsvikt uppdelat på två- fem på varandra följande dagar var fjärde vecka). Klar förbättring av kutan DM rapporterades i en randomiserad studie utformad för att utvärdera effektivitet av IVIG på myosit vid DM (65). Markant förbättring av hudmanifestationer sågs hos alla 8 patienter som behandlades med IVIG. Tyvärr rapporterades inte effekt på hudmanifestationer hos kontrollgruppen i denna studie. Ytterligare två retrospektiva studier stödjer användning av IVIG vid refraktär DM (15, 66, 67). I en nyligen publicerad randomiserad, placebokontrollerad studie inkluderande patienter med DM, som inte svarat på första linjens läkemedelsbehandling, sågs positiv effekt av högdos IVIG. Octagam® som användes i studien är nu godkänt av FDA för behandling av DM (3). Hos riskpatienter för trombos bör infusion ges i långsam takt och över 5 dagar.
Tredje linjens behandlingsval vid hudutslag
JAK hämmare: JAK-hämmare kan vara mycket effektiva mot kutan dermatomyosit (17). Tre patienter med kutan DM som inte hade svarat på flera DMARDs fick utmärkt klinisk respons på tofacitinib (5mgx2) efter fyra veckors behandling, och CDASI-score minskade hos samtliga patienter (68). I en open-label studie behandlades 10 patienter med refraktär DM med tofacitinib (11mgx1). Alla 10 patienter uppnådde en viss förbättring av sjukdomsaktiviteten efter 12 veckor och en signifikant minskning av CDASI-score observerades (från 28±15,4 vid studiestart till 9,5±8,5 vid vecka 12) (69). En öppen pilotstudie visade att alla patienter med initialt aktiv kutan sjukdomsaktivitet förbättrades med JAK-hämmare (Baricitinib och ruxolitinib) med signifikant minskad CDASI (70).
Rituximab (1000 mg x 2 givet med två veckors mellanrum): En posthoc analys av data från Rituximab vid myosit-studien antydde en gynnsam effekt på hudsymptom hos vuxna och barn med DM (16, 71). Gynnsam effekt av rituximab på hudmanifestationer har även rapporterats i en öppen studie där sju patienter med DM behandlades med fyra veckoinfusioner av rituximab. Alla fem patienter med hudmanifestationer i början av studien, uppvisade klar förbättring av hudsymptom. Dessutom inträffade håråterväxt hos två patienter med alopeci sekundärt till DM (72). I en annan öppen studie där rituximab visade viss effekt på myosit sågs däremot ingen effekt på hudsymptom (73).
Fjärde linjens behandlingsval vid hudutslag
Övriga terapier: Apremilast har i en fallrapport visat gynnsam effekt mot svår klåda som inte svarade på konventionell immunsuppressiv behandling (74). Ustekinumab visade drastisk förbättring i refraktär mekanikershänder hos en patient med antisyntetassyndrom (Fallrapport) (75). Anifrolumab, en human monoklonal Typ 1 Interferon receptor antikropp, registrerad för behandling av SLE, har rapporterats i enstaka fallrapporter ha effekt vid dermatomyosit och testas nu i en randomiserad klinisk prövning (76).
Calcinos
Dermatomyositpatienter kan utveckla calcinos, vilket kännetecknas av olösliga kalciumavlagringarna i huden. Cirka 20 % av vuxna patienter med dermatomyosit drabbas av calcinos, medan prevalensen är betydligt högre hos barn med juvenil dermatomyosit (JDM). Den exakta mekanismen bakom calcinos är fortfarande oklar, men förekomsten av MDA-5- och/eller NXP-2-antikroppar är starkt kopplad till dess utveckling vid dermatomyosit (77).
Rekommendationer för behandling av calcinos vid dermatomyosit baseras på begränsad evidens, eftersom stora kliniska studier saknas. Eftersom calcinos tros vara en följd av inflammation är tidig diagnos och effektiv kontroll av sjukdomsaktiviteten avgörande för att förebygga dess utveckling (77).
Kirurgisk excision kan övervägas för patienter med lokaliserad hudpåverkan. Intralesionell natriumtiosulfat (0,1–1 ml, 150-250 mg/ml, en gång per månad) har visat viss effekt. Kalciumkanalblockeraren diltiazem (2–4 mg/kg per dag) har visat effekt i fallserier, men är sedan 2024 avregistrerad i Sverige och kräver licens för att kunna förskrivas. När det gäller immunosuppressiva behandlingar har JAK-hämmare, TNF-hämmare och intravenöst immunglobulin (IVIG) visat sig förbättra calcinos både vid JDM och vuxen dermatomyosit (77, 78).
Behandling av subgrupp Inklusionskroppsmyosit (IBM)
Bakgrund
Inklusionskroppsmyosit (IBM) beskrevs första gången 1971 då man noterat att en del patienter som hade polymyosit inte svarade på behandling med kortison som förväntat. 1995 beskrevs de kliniska, labmässiga och muskelpatologiska fynden vid IBM av Griggs (79). Sjukdomen debuterar vanligen efter 45-årsåldern men kan undantagsvis debutera även i 30-årsåldern. Förhållandet avseende förekomst mellan män och kvinnor är 2:1. Man ser ofta ett asymmetriskt muskelengagemang med svaghet och atrofi (80). Sjukdomen utvecklas långsamt under förloppet av år vilket också leder till fördröjning av diagnos ofta dessvärre i flera år. Med tanke på att IBM debuterar i högre åldrar är det en viktig differentialdiagnos till ALS, motorneuronsjukdomar, neuropatier men även andra myositsjukdomar.
Sjukdomen involverar framför allt quadricepsmuskler, fingerflexorer och hos ca 2/3 även sväljningsmuskler. I sällsynta fall ses även engagemang av ansikts- och ögonmuskler och s k Bent Spine Syndrome (engagemang av paraspinala muskler). Analys av anti-cN1A kan vara av värde för diagnostiken. I muskelbiopsi ses inflammatoriska celler endomysialt och i klassiska fall s k rimmed vacuoles, proteinansamling, och tubulofilament. Tecken finns ofta på både degeneration av muskelfibrer och inflammatoriska cellinfiltrat.
Den idag mest använda definitionen av IBM presenterades 2011 och definierar: kliniskt patologiskt definierad IBM, kliniskt definierad IBM och sannolik IBM (81).
Den primära behandlingen av IBM är träning av drabbade muskler med hjälp av fysioterapeut och handfunktion med hjälp av arbetsterapeut. Vid dysfagi behöver man insatser av logoped och dietist. Med hjälp av fiberendoskopisk undersökning (FUS) kan striktur och hypertrofi påvisas av cricopharyngeusmuskeln och möjlighet till vidgning av muskeln på ÖNH-klinik förbättra sväljningsförmågan. Målet för interventionerna är att upprätthålla muskelstyrkan så länge som möjligt.
Farmakologisk behandling av inklusionskroppsmyosit
Kontrollerade studier med läkemedelsbehandling vid IBM är få. Ingen har hittills kunnat påvisa förbättring av muskelstyrkan. Några öppna studier har kunnat påvisa en minskad försämring av muskelstyrkan i jämförelse med det förväntade. Att notera, CK sjunker ofta under behandling med kortison och immundämpande läkemedel utan att klinisk förbättring sker. CK kan således inte vara en parameter för utvärdering av sjukdomen. Utvärdering bör istället ske med uppföljning av muskelstyrka/muskulär uthållighet (MMT/myositstatus).
KORTIKOSTEROIDER. Kortisonbehandling är kontroversiell vid IBM och ingen studie har påvisat en effekt vid IBM men kan ge viss förbättring i en del fall initialt (82-84). Prednisolonbehandling (0,5mg/kg) med nedtrappning med 10 mg per månad med utvärdering efter 3 månader kan vara ett sätt att starta läkemedelsbehandling men om ingen effekt ses avseende minskad progress av muskelsvaghet bör prednisolonbehandlingen avslutas (Figur 7). Långvarig kortisonbehandling utan effekt har enbart negativ påverkan på muskulaturen.
METOTREXATE (85) kan ha en viss effekt (10-20mg/vecka) och kan insättas som monoterapi om svar föreligger på kortisonbehandling. Alternativt kan man insätta azathioprin om kontraindikationer föreligger mot metotrexate.
IVIG hos vissa patienter med sväljningsbesvär (2g/kg kroppsvikt iv var fjärde vecka) kan ge en förbättring. Dessvärre tenderar förbättringen att avklinga om behandlingen avslutas.
Läkemedel som inte rekommenderas vid IBM i nuläget är TNF-hämmare (etanercept) (86), alemtuzumab/CAMPATH (87) eller betainterferon 1 a (88). Anakinra hade viss effekt mätt med International Myositis Assessment and Clinical Studies (IMACS) förbättringskriterier på enstaka patienter med IBM i en öppen studie (89).
Det finns i nuläget mycket lite som talar för att behandla med rituximab (90), abatacept (91) eller JAK-hämmare.
RAPAMYCIN (sirolimus), ett läkemedel som inhiberar mTOR och som används hos organtransplanterade patienter har i en studie (80), kunnat visa förbättring vad gäller sekundära endpoints såsom HAQ-DI, thigh fat fraction, forcerad vitalkapacitet och 6 minuters gångtest.
Träning vid Inklusionskroppsmyosit
Var god se avsnitt Träning vid myosit
Sammanfattning IBM
Sammanfattningsvis bör individuell bedömning göras inför behandling av patienter med IBM. Samtliga patienter med IBM bör erbjudas träning under instruktion av fysioterapeut. Läkemedelsbehandling med immunosuppression kan eventuellt prövas under en begränsad tid och särskilt till de patienter som har en kort sjukdomsduration och påtaglig inflammation i muskelbiopsin. Att tänka på är att en stabilisering av muskelstyrkan kan ses som en positiv effekt av behandlingen.
Behandling av subgrupp Immunmedierad nekrotiserande myopati (IMNM)
Bakgrund
Immunmedierad nekrotiserande myopati (IMNM) är en form av myopati som kännetecknas av en uttalat svår, nekrotiserande myosit, där sväljningssvårigheter inte sällan förekommer men ofta saknas lung- och hudengagemang (92). Prevalensdata visar på att tillståndet förekommer hos ca 2-3/100 000 invånare (93).
IMNM klassificeras oftast efter förekomst av de myositspecifika antikropparna anti-HMGCR och anti-SRP-ak, då majoriteten av patienterna har någon av dessa antikroppar, och endast ca 10% är seronegativa. Seronegativ sjukdom är oftare kopplad till underliggande malignitet (94-96). IMNM är en allvarlig sjukdom som kan ha en drastisk effekt på patienters livskvalitet om den inte handläggs snabbt (97). Flera riskfaktorer, såsom statinbehandling, malignitet och virala infektioner har kopplats till insjuknande (92, 98). Muskelbiopsi är viktig i diagnostiken och den domineras, till skillnad mot PM/DM/IBM, av muskelfibernekroser och regenererande muskelfibrer med minimala eller inga inflammatoriska fynd, (98, 99).
Den kliniska bilden vid IMNM präglas av en uttalad, bilateral, proximal symmetrisk muskelsvaghet som kan utvecklas under några veckor, eller långsammare över några månader (98). Karaktäristiskt för IMNM är mycket kraftigt förhöjda CK-nivåer (även jämfört med andra IIM), där medianvärden brukar ligga mellan 70-115 µkat/L, och följaktligen brukar myoglobin uppvisa fyrsiffrig stegring (100).
Förekomst av anti-HMGCR autoantikroppar är ofta associerad med statinbehandling, och är vanligare hos äldre patienter. Dessa kräver ofta lägre kortisondoser vid start, och är associerade med bättre behandlingssvar med IVIG-behandling än av andra DMARD.
Antikroppar mot SRP förekommer oftare hos yngre och denna form av IMNM kan svara på rituximab-behandling (94, 96, 97, 100).
Tyvärr saknas randomiserade kontrollerade studier för vägledning vid behandling av denna patientgrupp, utan behandlingsrekommendationer vilar huvudsakligen på fallrapporter och fallserier. Vid en internationell multidisciplinär workshop sammanfattades rekommendationer för behandling av patienter med IMNM (100).
Farmakologisk behandling
Anti-SRP positiv IMNM.
Första linjens behandlingsval vid anti-SRP positiv IMNM
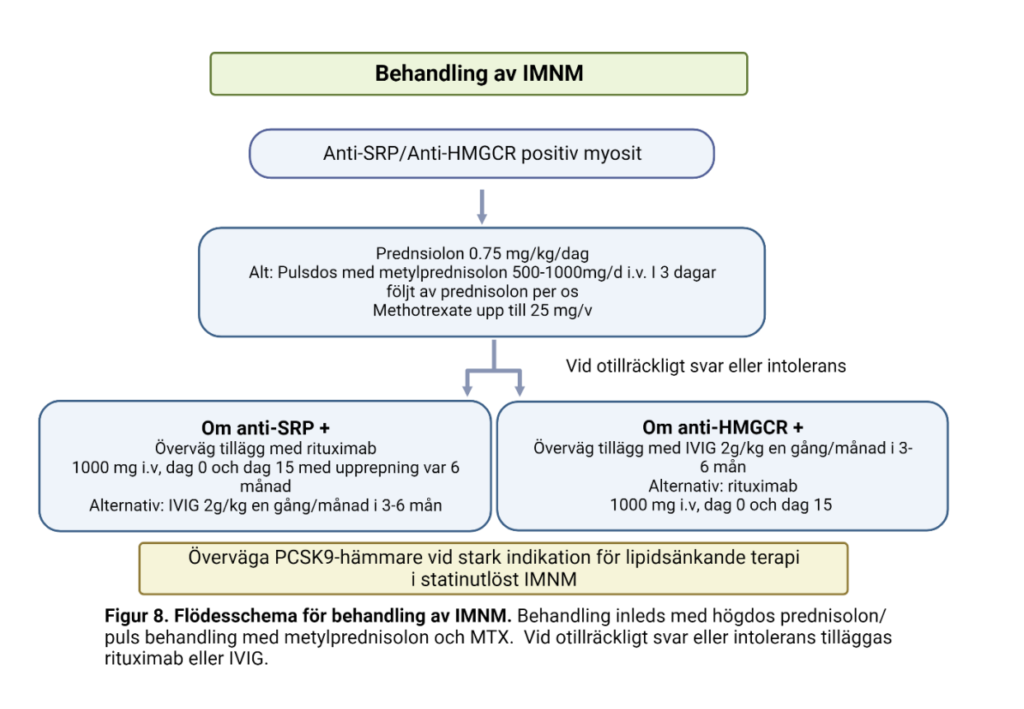
Vid förekomst av anti-SRP-ak med muskelsymtom rekommenderas induktionsbehandling med peroralt prednisolon alternativt i.v. pulsbehandling med metylprednisolon 500-1000 mg iv per dag i 3 dagar följt av prednisolon 0.75 mg/kg i kombination med metotrexate per os eller subkutant upp till 25 mg/vecka (Figur 8).
Andra linjens behandlingsval vid anti-SRP positiv IMNM
Vid utebliven förbättring efter 3 månader eller vid mycket uttalad muskelsvaghet rekommenderas tillägg med rituximab 1000 mg dag 0 och dag 15 och därefter 500-1000 mg var sjätte månad (92, 100).
Anti-HMGCR-positiv IMNM
Vid statinbehandling: sätt ut statiner.
Första linjens behandlingsval vid anti-HMGCR positiv IMNM
Vid anti-HMGCR positiv IMNM associerad med statinbehandling, kan man oftare ge lägre kortisondoser initialt än vid anti-SRP positiv sjukdom, exempelvis 0.5 mg/kg/dag. Kortison ges i kombination med 20-25 mg metotrexate veckovis som induktionsbehandling vid mindre svår presentation (Figur 8). Det finns även fallrapporter med induktionsbehandling med enbart högdos IVIG alternativt IVIG i kombination med metotrexate dvs utan kortison(101).
Andra linjens behandlingsval vid anti-HMGCR positiv IMNM
Vid svårare sjukdom eller dåligt svar, rekommenderas tillägg med IVIG (exempelvis 2g/kg månadsvis) i sex månader och därefter försök till utglesning/dosminskning (102).
Rituximab 1000 mg dag 0 och dag 15, och därefter 500-1000 mg var sjätte månad kan också övervägas (92, 100, 103).
Vid svår och behandlingsrefraktär IMNM som recidiverar trots ovanstående, kan man överväga plasmabyte. Evidensen är dock skral (104).
Behandling bör pågå i minst 2 års tid varefter de-eskalering kan övervägas utifrån den kliniska bilden. Någon evidens avseende optimal uppföljning och frekvens av återbesök saknas och bör anpassas till den individuella patienten. Målet med behandlingen bör vara maximering av muskelstyrka, normalisering av CK-/myoglobinstegring och minimering av läkemedelsbiverkan (105).
Lipidsänkande terapi
För de patienter som efter insjuknande i statinutlöst IMNM där det finns stark indikation för fortsatt lipidsänkande terapi, kan man i samråd med kardiolog överväga insättning av PCSK9-hämmare (106). Dessa har i större översikter inte förknippats med muskelbiverkan (107, 108). Data vid IMNM är begränsade, men det finns en växande klinisk erfarenhet av att PCSK9-hämmare inte förefaller utlösa återfall/försämring hos patienter som tidigare drabbats av statin-relaterad IMNM (109).
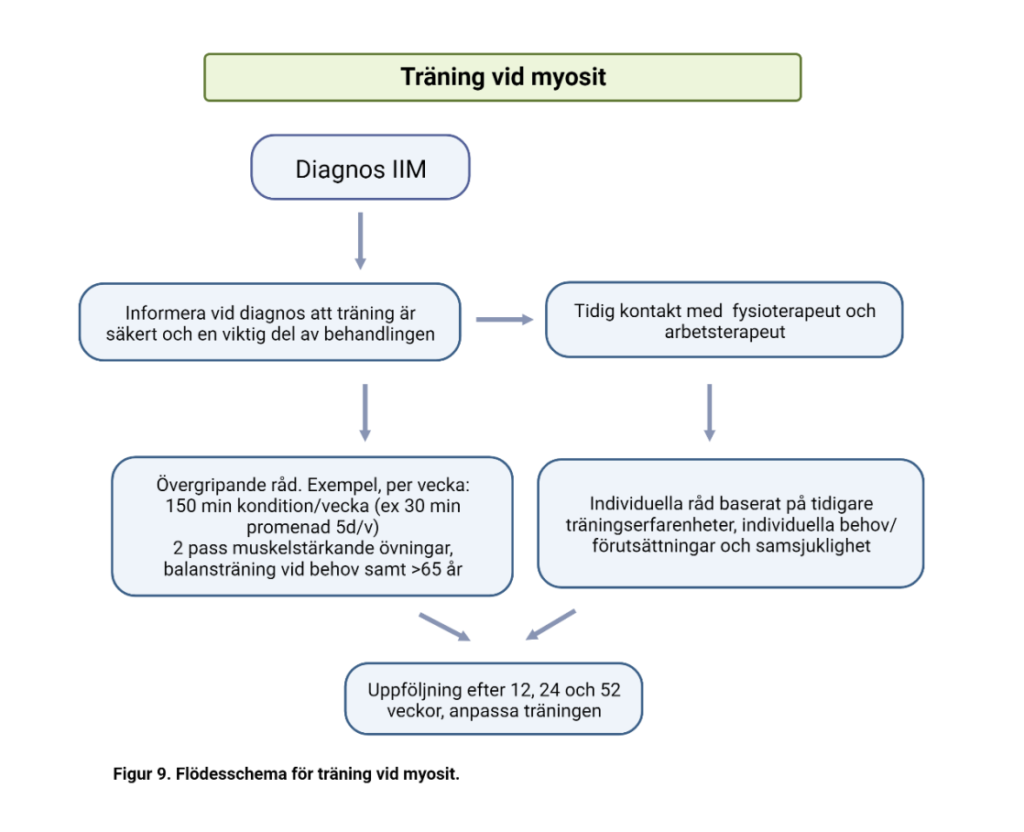
Rekommendationer om träning vid myosit
Bakgrund
Avseende träning vid idiopatisk inflammatorisk myopati (IIM), eller myosit, så är det vetenskapliga underlaget begränsat. Det finns få randomiserade kontrollerade studier. De flesta studier är små (n= 1 – 57) och med kort duration (median 12 veckor, range 6-24). Flest studier inkluderar patienter med polymyosit och dermatomyosit men även en del studier på inklusionskroppsmyosit (IBM). Endast enstaka studier har utvärderat effekt av träning vid antisyntetassyndrom (ASyS) och immunmedierad nekrotiserande myopati (IMNM) (110-112) samt enstaka studier som studerat träningseffekter vid nydebuterad/aktiv sjukdom (113-117). Vi kan dock anta att ASyS och IMNM varit inkluderade i polymyosit enligt äldre diagnoskriterier. Många olika former av träning har använts i de olika studierna, kondition (exempelvis gång, löpband, cykling), kondition + styrketräning eller bara styrketräning. Både träning under ledning av fysioterapeut och hembaserad träning har studerats. Uppvärmning och stretch har varit en del av interventionen i många studier, men beskrivs inte så utförligt. En rad olika utfallsmått, de flesta för att mäta kondition och muskelfunktion har använts. Ofta är träningsdosen ospecificerad när det gäller intensitet (% av maximal kapacitet) och duration per pass, men studier publicerade under de senaste 10-15 åren beskriver träningen mer specifikt enligt Frekvens (antal pass per vecka), Intensitet (% av VO2peak/% av maximal styrka), Tid per träningspass och Tid (duration i veckor) (FITT).
Säkerhet
Sammantaget visade en absolut majoritet av studierna förbättrad muskelfunktion och oförändrad eller i vissa fall minskad sjukdomsaktivitet (mätt på olika sätt såsom CK, MMT8, HAQ, ”flare”, SR, CRP, MITAX och olika mått i muskelbiopsier och/eller magnetkamera) (110-113, 116-119). Endast en studie som utvärderade effekt av submaximal styrketräning vid strypt blodflöde vid IBM rapporterade en gränssignifikant ökning i CK-nivåer i serum, men signifikant skillnad jämfört med kontrollgruppen. Dock indikerade patientens och läkarens bedömning av sjukdomsaktivitet och skada att träningen inte resulterade i ökad sjukdomsaktivitet (120).
Effekt av träning vid PM, DM, ASyS, IMNM
Över lag förbättrade konditionsträning den aeroba kapaciteten (121-125) medan styrketräning gav starkare muskler (118, 126). En del studier kombinerade konditionsträning med styrketräning, ofta med förbättring av både aerob kapacitet och muskelstyrka (110, 127, 128). Intensiv konditionsträning på 70 % av maximal kapacitet och i kombination med muskulär uthållighetsträning (30–40 repetitioner till muskulär utmattning) som utfördes 60 minuter, 3 gånger per vecka ledde till minskad sjukdomsaktivitet, minskad inflammation i muskelvävnaden samt förbättrade muskelns aeroba kapacitet (ökad mitokondrieenzymaktivitet, ökat antal kapillärer) hos personer med stabil, inflammatoriskt lågaktiv myosit (121-123). Olika hemträningsprogram där styrkeövningar kombinerades med aerob träning (gång) verkade även de ha en positiv effekt på muskelfunktionen (114, 129, 130). Tillägg av konditionsträning till ”rehabiliteringsträning” har i en retrospektiv studie på PM och DM visat på tilläggseffekter både avseende både muskelstyrka (MMT-8) och muskeluthållighet (FI-2) samt välbefinnande (SF-36) (131). 16 veckors styrketräning på 10 VRM (ca 70% av maximal styrka) resulterade i signifikant förbättrad livskvalitet (fysiska domäner), muskelstyrka och muskulär uthållighet jämfört med träning som vanligtvis förskrivs hos patienter med etablerad myosit (126). Uppföljning efter ett år visade kvarvarande effekter på muskeluthållighet mätt med functional index 3 (126). Vidare minskade patientens globala skattning av sjukdomsaktivitet jämfört med kontrollgruppen. 24 veckors funktionell styrketräning minskade depression, förbättrade muskelfunktion och minskade sjukdomsaktivitet jämfört med en icke tränande kontrollgrupp (112).
En pilotstudie har utvärderat handträning 3 gånger per vecka under 12 veckor hos patienter med PM och DM. De utförde 8 olika handträningsövningar med hjälp av ”motståndsdegar” med ökande antal repetitioner var 4:e vecka. Studien visade att programmet var genomförbart med vissa individuella förbättringar av handfunktionen men att motståndet troligen skulle behöva ökas för att uppnå bättre effekt (132)
Ett hemträningsprogram är utvärderat även hos patienter med nydebuterad, aktiv IIM. Hemträning, ca 10 repetitioner i 8 muskelgrupper, samt promenader 5 dagar per vecka tolereras väl om träningen anpassas individuellt (113). Förutom effekter på muskelfunktion finns det även studier som visar på förbättrad livskvalitet och mindre trötthet/fatigue efter träning (113, 114, 122, 126, 133).
Effekt av träning vid IBM
En randomiserad kontrollerad studie (RCT) utvärderade träning av lårmusklerna under strypt blodtillförsel (blood flow restriction, BFR) hos patienter med IBM. Studien visade att submaximal styrketräning som utfördes två gånger per vecka i 12 veckor under 70 % strypt blodflöde bidrog till bibehållen muskelstyrka i quadriceps jämfört med en statistiskt säkerställd försämring i styrka på nästan 10 % hos kontrollgruppen (134). En annan RCT visade att 12 veckors kombinerad konditions- och styrketräning förbättrade aerob kapacitet hos personer med IBM jämfört med kontrollgruppen (135). Två studier har utvärderat intensiv hemträning för personer med IBM. En av studierna indikerar att hemträning som utförs två gånger per dag i 16 veckor kan förbättra muskelstyrka och förbättra funktion såsom kort gångsträcka och förmåga att resa sig från en stol (136). Tio års klinisk erfarenhet indikerar att de flesta patienter tolererar denna hemträning väl, förutsatt individuell anpassning, framför allt som en introduktion till träning att sedan bygga vidare på. Vidare kan aerob träning på motionscykel 3 gånger per vecka (80 % av maximal kapacitet) i kombination med samma hemträningsprogram förbättra konditionen hos personer med IBM. Dock sågs ingen förbättring i muskelstyrka (137). En annan studie på patienter med IBM-kunde inte påvisa någon förbättrad muskelstyrka efter 12 veckors hemträning, men inte heller någon försämring (119).
Träningsrekommendationer
Med bakgrund av den begränsade litteraturen är det svårt att ge ingående rekommendationer kring duration av träningspass, intensitet, typ av träning etc. Det finns dock tillräckligt med vetenskapligt underlag som stödjer att träning är säkert. Konditionsträning ökar uthålligheten och styrketräning stärker musklerna, även i denna patientgrupp. Med avseende på patienter med inflammatoriskt aktiv sjukdom som står på höga doser kortison eller har annan samsjuklighet är data så begränsade att det är svårt att ge rekommendation på gruppnivå. Det är viktigt att komma igång med träning tidigt för att potentiellt minska biverkningar av medicinsk behandling och kanske också själva sjukdomen, samt att stimulera till en långvarig fysiskt aktiv livsstil hos personer som lever med myosit (Figur 9). Studier och många års klinisk erfarenhet indikerar att hemträning 5 dagar per vecka tolereras väl av personer med nydebuterad/aktiv myosit, förutsatt att träningen anpassas individuellt till patientens muskelfunktion, trötthet och smärta. Här bör individuell bedömning göras i samråd med fysioterapeut och behandlande läkare, vilket också gäller patienter med allvarliga hjärt- och lungengagemang. Man bör även komma ihåg att träning kan ge en fysiologisk CK-stegring. En systematisk litteraturöversikt visar att CK-värden når sin topp efter 24 till 48 timmar beroende på intensitet och duration av träningspass (138). Det är därför viktigt att informera patienten att vänta minst 48 men gärna 72 timmar efter träning innan de lämnar prover.
Som vägledning för rekommendationer finns det en systematisk översiktsartikel som presenterar evidensbaserade rekommendationer för patienter med myosit baserade på 3 RCT och en systematisk översiktsartikel (med patientrelaterade utfallsmått). För att öka den aeroba kapaciteten rekommenderas 90-180 minuter (på en intensitet av 60-70 % av max), fördelat på 3 dagar per vecka (139). Detta är i linje med de rekommendationer som finns för allmänheten avseende fysisk aktivitet (minst 150min/vecka måttlig intensitet eller minst 75 minuter/vecka på hög intensitet). I den ovannämnda systematiska översikten ges inga specifika rekommendationer för styrketräning då evidens saknas. I den generella populationen rekommenderas två muskelstärkande pass per vecka och det kan fungera som ett riktmärke även vid inflammatoriska myopatier (i väntan på bättre evidens). Vid behov och hos individer över 65 år rekommenderas även balansträning (www.fyss.se).
Flödesscheman
Bilder/flödesscheman Myosit




Referenser
- usefulness of myositis specific antibody. Brain 139, 2131-2135 (2016).
- Y. Allenbach, O. Benveniste, Peculiar clinicopathological features of immune-mediated necrotizing myopathies. Current opinion in rheumatology 30, 655-663 (2018).
- C. D. Kassardjian, V. A. Lennon, N. B. Alfugham, M. Mahler, M. Milone, Clinical Features and Treatment Outcomes of Necrotizing Autoimmune Myopathy. JAMA Neurol 72, 996-1003 (2015).
- J. A. Day, V. Limaye, Immune-mediated necrotising myopathy: A critical review of current concepts. Semin Arthritis Rheum 49, 420-429 (2019).
- Y. Allenbach, A. L. Mammen, O. Benveniste, W. Stenzel, G. Immune-Mediated Necrotizing Myopathies Working, 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord 28, 87-99 (2018).
- J. Lim et al., Intravenous immunoglobulins as first-line treatment in idiopathic inflammatory myopathies: a pilot study. Rheumatology (Oxford) 60, 1784-1792 (2021).
- J. Suh, A. A. Amato, Effectiveness and Safety of IVIG for the Treatment of HMGCR Immune-Mediated Necrotizing Myopathy. Muscle Nerve 71, 392-397 (2025).
- A. L. Mammen, E. Tiniakou, Intravenous Immune Globulin for Statin-Triggered Autoimmune Myopathy. N Engl J Med 373, 1680-1682 (2015).
- R. L. Kruse et al., Therapeutic plasma exchange for the treatment of refractory necrotizing autoimmune myopathy. J Clin Apher 37, 253-262 (2022).
- E. Weeding, E. Tiniakou, Therapeutic management of immune-mediated necrotizing I. E. Lundberg et al., Idiopathic inflammatory myopathies. Nat Rev Dis Primers 7, 86 (2021).
- A. G. S. Oldroyd et al., British Society for Rheumatology guideline on management of paediatric, adolescent and adult patients with idiopathic inflammatory myopathy. Rheumatology (Oxford) 61, 1760-1768 (2022).
- F. W. Miller et al., Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 40, 1262-1273 (2001).
- R. Aggarwal et al., 2016 American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis 76, 792-801 (2017).
- H. Alexanderson et al., Functional index-2: Validity and reliability of a disease-specific measure of impairment in patients with polymyositis and dermatomyositis. Arthritis Rheum 55, 114-122 (2006).
- J. B. Lilleker et al., Using serum troponins to screen for cardiac involvement and assess disease activity in the idiopathic inflammatory myopathies. Rheumatology (Oxford) 57, 1041-1046 (2018).
- V. M. Ferreira et al., Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J Am Coll Cardiol 72, 3158-3176 (2018).
- A. Rosenbohm et al., Early diagnosis of cardiac involvement in idiopathic inflammatory myopathy by cardiac magnetic resonance tomography. J Neurol 262, 949-956 (2015).
- M. T. de Moraes, F. H. de Souza, T. B. de Barros, S. K. Shinjo, Analysis of metabolic syndrome in adult dermatomyositis with a focus on cardiovascular disease. Arthritis Care Res (Hoboken) 65, 793-799 (2013).
- P. A. O. Araujo, M. G. Silva, E. F. Borba, S. K. Shinjo, High prevalence of metabolic syndrome in antisynthetase syndrome. Clin Exp Rheumatol 36, 241-247 (2018).
- F. H. de Souza, S. K. Shinjo, The high prevalence of metabolic syndrome in polymyositis. Clin Exp Rheumatol 32, 82-87 (2014).
- B. Hanna et al., Cardiovascular risk and cardiac involvement in idiopathic inflammatory myopathies: insights from a cross-sectional Swedish single-centre study. Scandinavian journal of rheumatology 54, 272-281 (2025).
- C. Parraga Prieto et al., Similar risk of cardiovascular events in idiopathic inflammatory myopathy and rheumatoid arthritis in the first 5 years after diagnosis. Clin Rheumatol 40, 231-238 (2021).
- A. G. S. Oldroyd et al., International Guideline for Idiopathic Inflammatory Myopathy-Associated Cancer Screening: an International Myositis Assessment and Clinical Studies Group (IMACS) initiative. Nat Rev Rheumatol 19, 805-817 (2023).
- R. Aggarwal et al., Trial of Intravenous Immune Globulin in Dermatomyositis. N Engl J Med 387, 1264-1278 (2022).
- C. V. Oddis et al., Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 65, 314-324 (2013).
- J. J. Paik et al., Use of Janus kinase inhibitors in dermatomyositis: a systematic literature review. Clin Exp Rheumatol 41, 348-358 (2023).
- M. Beckett et al., Tofacitinib therapy in refractory inflammatory myositis: a retrospective cohort study of 41 patients. Rheumatology (Oxford) 10.1093/rheumatology/kead404 (2023).
- F. Muller et al., CD19 CAR T-Cell Therapy in Autoimmune Disease – A Case Series with Follow-up. N Engl J Med 390, 687-700 (2024).
- I. Marie et al., Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol 28, 2230-2237 (2001).
- E. Trallero-Araguas et al., Clinical manifestations and long-term outcome of anti-Jo1 antisynthetase patients in a large cohort of Spanish patients from the GEAS-IIM group. Semin Arthritis Rheum 46, 225-231 (2016).
- S. R. Johnson et al., 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) Guideline for the Screening and Monitoring of Interstitial Lung Disease in People with Systemic Autoimmune Rheumatic Diseases. Arthritis Rheumatol 76, 1201-1213 (2024).
- I. Marie et al., Interstitial lung disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Care Res (Hoboken) 65, 800-808 (2013).
- K. M. Antoniou et al., ERS/EULAR clinical practice guidelines for connective tissue diseases associated interstitial lung disease. Ann Rheum Dis 10.1016/j.ard.2025.08.021 (2025).
- T. Fujisawa et al., Prednisolone and tacrolimus versus prednisolone and cyclosporin A to treat polymyositis/dermatomyositis-associated ILD: A randomized, open-label trial. Respirology 26, 370-377 (2021).
- R. W. Hallowell, J. J. Paik, Myositis-associated interstitial lung disease: a comprehensive approach to diagnosis and management. Clin Exp Rheumatol 40, 373-383 (2022).
- J. A. Huapaya et al., Long-term treatment with human immunoglobulin for antisynthetase syndrome-associated interstitial lung disease. Respir Med 154, 6-11 (2019).
- I. C. Mira-Avendano et al., A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir Med 107, 890-896 (2013).
- N. Sharma, M. S. Putman, R. Vij, M. E. Strek, A. Dua, Myositis-associated Interstitial Lung Disease: Predictors of Failure of Conventional Treatment and Response to Tacrolimus in a US Cohort. J Rheumatol 44, 1612-1618 (2017).
- F. Ingegnoli et al., Interstitial lung disease outcomes by high-resolution computed tomography (HRCT) in Anti-Jo1 antibody-positive polymyositis patients: a single centre study and review of the literature. Autoimmun Rev 11, 335-340 (2012).
- T. Kurita et al., The efficacy of tacrolimus in patients with interstitial lung diseases complicated with polymyositis or dermatomyositis. Rheumatology (Oxford) 54, 39-44 (2015).
- M. R. Wilkes, S. M. Sereika, N. Fertig, M. R. Lucas, C. V. Oddis, Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 52, 2439-2446 (2005).
- T. M. Maher et al., Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): a double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir Med 11, 45-54 (2023).
- K. Kouranloo et al., Management and outcomes of interstitial lung disease associated with anti-synthetase syndrome: A systematic literature review. Rheumatology (Oxford) 10.1093/rheumatology/keae403 (2024).
- R. Aggarwal et al., Abatacept for the treatment of myositis-associated interstitial lung disease (ATtackMy-ILD). Rheumatology (Oxford) 10.1093/rheumatology/keaf218 (2025).
- V. Langlois et al., Rituximab and Cyclophosphamide in Antisynthetase Syndrome-related Interstitial Lung Disease: An Observational Retrospective Study. J Rheumatol 47, 1678-1686 (2020).
- Y. Zhao et al., Effect size of rituximab on pulmonary function in the treatment of connective-tissue disease-related interstitial lung disease: a systematic review and meta-analysis. Respir Res 23, 164 (2022).
- H. Tsuji et al., Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy With High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol 72, 488-498 (2020).
- H. You et al., Triple-combination therapy did not improve prognosis in anti-MDA5 positive dermatomyositis: a multicentre longitudinal cohort study. Clin Exp Rheumatol 43, 251-259 (2025).
- C. He, W. Li, Q. Xie, G. Yin, Rituximab in the Treatment of Interstitial Lung Diseases Related to Anti-Melanoma Differentiation-Associated Gene 5 Dermatomyositis: A Systematic Review. Front Immunol 12, 820163 (2021).
- M. McPherson, S. Economidou, A. Liampas, P. Zis, K. Parperis, Management of MDA-5 antibody positive clinically amyopathic dermatomyositis associated interstitial lung disease: A systematic review. Semin Arthritis Rheum 53, 151959 (2022).
- B. Tangborwornweerakul, N. Phutthinart, S. Disayabutr, W. Katchamart, One-year survival benefit of plasma exchange in idiopathic inflammatory myositis patients with progressive interstitial lung disease-a systemic review and meta-analysis. Semin Arthritis Rheum 69, 152564 (2024).
- T. Shirai, T. Machiyama, H. Sato, T. Ishii, H. Fujii, Intensive induction therapy combining tofacitinib, rituximab and plasma exchange in severe anti-melanoma differentiation-associatedprotein-5 antibody-positive dermatomyositis. Clin Exp Rheumatol 41, 291-300 (2023).
- W. Wu et al., Effectiveness and safety of tofacitinib versus calcineurin inhibitor in interstitial lung disease secondary to anti-MDA5-positive dermatomyositis: a multicentre cohort study. Eur Respir J 65 (2025).
- U. Lindgren, R. Pullerits, C. Lindberg, A. Oldfors, Epidemiology, Survival, and Clinical Characteristics of Inclusion Body Myositis. Ann Neurol 92, 201-212 (2022).
- P. Cherin et al., Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology 58, 326 (2002).
- M. C. Dalakas, Intravenous immune globulin therapy for neurologic diseases. Ann Intern Med 126, 721-730 (1997).
- M. C. Dalakas, Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology 51, S2-8 (1998).
- C. Dobloug, R. Walle-Hansen, J. T. Gran, O. Molberg, Long-term follow-up of sporadic inclusion body myositis treated with intravenous immunoglobulin: a retrospective study of 16 patients. Clin Exp Rheumatol 30, 838-842 (2012).
- K. Pars et al., Subcutaneous immunoglobulin treatment of inclusion-body myositis stabilizes dysphagia. Muscle Nerve 48, 838-839 (2013).
- R. B. Ramachandran, M. Swash, Pharyngeal Dysphagia in dermatomyositis: responsive to cyclophosphamide. J Clin Neuromuscul Dis 5, 166-167 (2004).
- L. Dourmishev, H. Meffert, H. Piazena, Dermatomyositis: comparative studies of cutaneous photosensitivity in lupus erythematosus and normal subjects. Photodermatol Photoimmunol Photomed 20, 230-234 (2004).
- W. K. Cheong, G. R. Hughes, P. G. Norris, J. L. Hawk, Cutaneous photosensitivity in dermatomyositis. Br J Dermatol 131, 205-208 (1994).
- J. P. Callen, R. L. Wortmann, Dermatomyositis. Clin Dermatol 24, 363-373 (2006).
- R. D. Quain, V. P. Werth, Management of cutaneous dermatomyositis: current therapeutic options. Am J Clin Dermatol 7, 341-351 (2006).
- T. Yoshimasu, T. Ohtani, T. Sakamoto, A. Oshima, F. Furukawa, Topical FK506 (tacrolimus) therapy for facial erythematous lesions of cutaneous lupus erythematosus and dermatomyositis. Eur J Dermatol 12, 50-52 (2002).
- C. B. Hollar, J. L. Jorizzo, Topical tacrolimus 0.1% ointment for refractory skin disease in dermatomyositis: a pilot study. J Dermatolog Treat 15, 35-39 (2004).
- J. Pinard et al., Systemic Treatment for Clinically Amyopathic Dermatomyositis at 4 Tertiary Care Centers. JAMA Dermatol 155, 494-496 (2019).
- R. D. Sontheimer, The management of dermatomyositis: current treatment options. Expert Opin Pharmacother 5, 1083-1099 (2004).
- M. L. Jewell, D. P. McCauliffe, Patients with cutaneous lupus erythematosus who smoke are less responsive to antimalarial treatment. J Am Acad Dermatol 42, 983-987 (2000).
- T. Hornung, A. Ko, T. Tuting, T. Bieber, J. Wenzel, Efficacy of low-dose methotrexate in the treatment of dermatomyositis skin lesions. Clin Exp Dermatol 37, 139-142 (2012).
- J. S. Kasteler, J. P. Callen, Low-dose methotrexate administered weekly is an effective corticosteroid-sparing agent for the treatment of the cutaneous manifestations of dermatomyositis. J Am Acad Dermatol 36, 67-71 (1997).
- P. W. Wolstencroft, L. Chung, S. Li, L. Casciola-Rosen, D. F. Fiorentino, Factors Associated With Clinical Remission of Skin Disease in Dermatomyositis. JAMA Dermatol 154, 44-51 (2018).
- J. C. Edge, J. D. Outland, J. R. Dempsey, J. P. Callen, Mycophenolate mofetil as an effective corticosteroid-sparing therapy for recalcitrant dermatomyositis. Arch Dermatol 142, 65-69 (2006).
- M. C. Dalakas et al., A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 329, 1993-2000 (1993).
- A. N. Femia et al., Intravenous immunoglobulin for refractory cutaneous dermatomyositis: a retrospective analysis from an academic medical center. J Am Acad Dermatol 69, 654-657 (2013).
- T. Bounfour et al., Clinical efficacy of intravenous immunoglobulins for the treatment of dermatomyositis skin lesions without muscle disease. J Eur Acad Dermatol Venereol 28, 1150-1157 (2014).
- D. J. Kurtzman et al., Tofacitinib Citrate for Refractory Cutaneous Dermatomyositis: An Alternative Treatment. JAMA Dermatol 152, 944-945 (2016).
- J. J. Paik et al., Study of Tofacitinib in Refractory Dermatomyositis: An Open-Label Pilot Study of Ten Patients. Arthritis Rheumatol 73, 858-865 (2021).
- O. Landon-Cardinal et al., JAK inhibitors for the treatment of adult dermatomyositis: A pilot study. J Am Acad Dermatol 88, 924-926 (2023).
- R. Aggarwal et al., Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology (Oxford) 56, 247-254 (2017).
- T. D. Levine, Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum 52, 601-607 (2005).
- L. Chung, M. C. Genovese, D. F. Fiorentino, A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol 143, 763-767 (2007).
- D. Charlton et al., Refractory Cutaneous Dermatomyositis With Severe Scalp Pruritus Responsive to Apremilast. J Clin Rheumatol 27, S561-S562 (2021).
- I. Pinal-Fernandez, C. T. Kroodsma, A. L. Mammen, Successful treatment of refractory mechanic’s hands with ustekinumab in a patient with the antisynthetase syndrome. Rheumatology (Oxford) 58, 1307-1308 (2019).
- A. Claudio-Oliva et al., Anifrolumab use in refractory dermatomyositis: a case report and literature review. Clin Exp Dermatol 10.1093/ced/llae476 (2024).
- S. Davuluri, L. Chung, C. Lood, Calcinosis in dermatomyositis. Current opinion in rheumatology 36, 453-458 (2024).
- S. Davuluri, B. Duvvuri, C. Lood, S. Faghihi-Kashani, L. Chung, Calcinosis in dermatomyositis: Origins and possible therapeutic avenues. Best Pract Res Clin Rheumatol 36, 101768 (2022).
- R. C. Griggs et al., Inclusion body myositis and myopathies. Ann Neurol 38, 705-713 (1995).
- M. Pawlitzki, C. Nelke, M. Korsen, S. G. Meuth, T. Ruck, Sirolimus leads to rapid and sustained clinical improvement of motor deficits in a patient with inclusion body myositis. Eur J Neurol 29, 1284-1287 (2022).
- M. R. Rose, E. I. W. Group, 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 23, 1044-1055 (2013).
- R. L. Leff, F. W. Miller, J. Hicks, D. D. Fraser, P. H. Plotz, The treatment of inclusion body myositis: a retrospective review and a randomized, prospective trial of immunosuppressive therapy. Medicine (Baltimore) 72, 225-235 (1993).
- B. P. Lotz, A. G. Engel, H. Nishino, J. C. Stevens, W. J. Litchy, Inclusion body myositis. Observations in 40 patients. Brain 112 ( Pt 3), 727-747 (1989).
- M. E. Sayers, S. M. Chou, L. H. Calabrese, Inclusion body myositis: analysis of 32 cases. J Rheumatol 19, 1385-1389 (1992).
- U. A. Badrising et al., Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol 51, 369-372 (2002).
- R. J. Barohn et al., Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology 66, S123-124 (2006).
- M. C. Dalakas et al., Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain 132, 1536-1544 (2009).
- G. Muscle Study, Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology 63, 718-720 (2004).
- M. Zong et al., Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann Rheum Dis 73, 913-920 (2014).
- S. Vordenbaumen, E. Neuen-Jacob, J. Richter, M. Schneider, Inclusion body myositis in a patient with long standing rheumatoid arthritis treated with anti-TNFalpha and rituximab. Clin Rheumatol 29, 555-558 (2010).
- E. A. Lindberg C, Edofsson U, Hammaren E. , Experience with abatacept treatment in sporadic inclusion body myositis. Neuromuscular Disorders 25 (2015).
- I. Pinal-Fernandez, M. Casal-Dominguez, A. L. Mammen, Immune-Mediated Necrotizing Myopathy. Curr Rheumatol Rep 20, 21 (2018).
- A. Meyer et al., Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford) 54, 50-63 (2015).
- J. Lim et al., Seronegative patients form a distinctive subgroup of immune-mediated necrotizing myopathy. Neurol Neuroimmunol Neuroinflamm 6, e513 (2019).
- J. Schmidt, Current Classification and Management of Inflammatory Myopathies. J Neuromuscul Dis 5, 109-129 (2018).
- Y. Allenbach et al., High risk of cancer in autoimmune necrotizing myopathies: myositis. Curr Treatm Opt Rheumatol 7, 150-160 (2021).
- M. C. Mizus, E. Tiniakou, Lipid-lowering Therapies in Myositis. Curr Rheumatol Rep 22, 70 (2020).
- X. L. Zhang et al., Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med 13, 123 (2015).
- S. E. Nissen et al., Efficacy and Tolerability of Evolocumab vs Ezetimibe in Patients With Muscle-Related Statin Intolerance: The GAUSS-3 Randomized Clinical Trial. JAMA 315, 1580-1590 (2016).
- E. Tiniakou, E. Rivera, A. L. Mammen, L. Christopher-Stine, Use of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors in Statin-Associated Immune-Mediated Necrotizing Myopathy: A Case Series. Arthritis Rheumatol 71, 1723-1726 (2019).
- J. M. de Souza et al., Feasibility, safety and efficacy of exercise training in immune-mediated necrotising myopathies: a quasi-experimental prospective study. Clin Exp Rheumatol 37, 235-241 (2019).
- M. Nemec et al., Altered dynamics of lipid metabolism in muscle cells from patients with idiopathic inflammatory myopathy is ameliorated by 6 months of training. J Physiol 599, 207-229 (2021).
- M. Spiritovic et al., The effect of a 24-week training focused on activities of daily living, muscle strengthening, and stability in idiopathic inflammatory myopathies: a monocentric controlled study with follow-up. Arthritis Res Ther 23, 173 (2021).
- H. Alexanderson et al., Resistive home exercise in patients with recent-onset polymyositis and dermatomyositis — a randomized controlled single-blinded study with a 2-year followup. J Rheumatol 41, 1124-1132 (2014).
- H. Alexanderson, C. H. Stenstrom, G. Jenner, I. Lundberg, The safety of a resistive home exercise program in patients with recent onset active polymyositis or dermatomyositis. Scandinavian journal of rheumatology 29, 295-301 (2000).
- A. Escalante, L. Miller, T. D. Beardmore, Resistive exercise in the rehabilitation of polymyositis/dermatomyositis. J Rheumatol 20, 1340-1344 (1993).
- M. A. Mattar et al., Safety and possible effects of low-intensity resistance training associated with partial blood flow restriction in polymyositis and dermatomyositis. Arthritis Res Ther 16, 473 (2014).
- C. Varju, E. Petho, R. Kutas, L. Czirjak, The effect of physical exercise following acute disease exacerbation in patients with dermato/polymyositis. Clin Rehabil 17, 83-87 (2003).
- H. Alexanderson, M. Dastmalchi, M. Esbjornsson-Liljedahl, C. H. Opava, I. E. Lundberg, Benefits of intensive resistance training in patients with chronic polymyositis or dermatomyositis. Arthritis Rheum 57, 768-777 (2007).
- S. Arnardottir, H. Alexanderson, I. E. Lundberg, K. Borg, Sporadic inclusion body myositis: pilot study on the effects of a home exercise program on muscle function, histopathology and inflammatory reaction. J Rehabil Med 35, 31-35 (2003).
- A. N. Jorgensen, P. Aagaard, U. Frandsen, E. Boyle, L. P. Diederichsen, Blood-flow restricted resistance training in patients with sporadic inclusion body myositis: a randomized controlled trial. Scandinavian journal of rheumatology 47, 400-409 (2018).
- L. Alemo Munters et al., Improvement in health and possible reduction in disease activity using endurance exercise in patients with established polymyositis and dermatomyositis: a multicenter randomized controlled trial with a 1-year open extension followup. Arthritis Care Res (Hoboken) 65, 1959-1968 (2013).
- L. Alemo Munters et al., Improved exercise performance and increased aerobic capacity after endurance training of patients with stable polymyositis and dermatomyositis. Arthritis Res Ther 15, R83 (2013).
- L. A. Munters et al., Endurance Exercise Improves Molecular Pathways of Aerobic Metabolism in Patients With Myositis. Arthritis Rheumatol 68, 1738-1750 (2016).
- G. F. Wiesinger et al., Improvement of physical fitness and muscle strength in polymyositis/dermatomyositis patients by a training programme. Br J Rheumatol 37, 196-200 (1998).
- G. F. Wiesinger et al., Benefit of 6 months long-term physical training in polymyositis/dermatomyositis patients. Br J Rheumatol 37, 1338-1342 (1998).
- K. Y. Jensen et al., High-intensity resistance training improves quality of life, muscle endurance and strength in patients with myositis: a randomised controlled trial. Rheumatol Int 44, 1909-1921 (2024).
- D. S. de Oliveira et al., Exercise training attenuates insulin resistance and improves beta-cell function in patients with systemic autoimmune myopathies: a pilot study. Clin Rheumatol 38, 3435-3442 (2019).
- D. S. de Oliveira et al., Exercise training attenuates skeletal muscle fat infiltration and improves insulin pathway of patients with immune-mediated necrotizing myopathies and dermatomyositis. Arch Rheumatol 38, 189-199 (2023).
- H. Alexanderson, C. H. Stenstrom, I. Lundberg, Safety of a home exercise programme in patients with polymyositis and dermatomyositis: a pilot study. Rheumatology (Oxford) 38, 608-611 (1999).
- M. Dastmalchi et al., Effect of physical training on the proportion of slow-twitch type I muscle fibers, a novel nonimmune-mediated mechanism for muscle impairment in polymyositis or dermatomyositis. Arthritis Rheum 57, 1303-1310 (2007).
- G. Zhang et al., Effects of Conventional Rehabilitative and Aerobic Training in Patients with Idiopathic Inflammatory Myopathy. Rheumatol Immunol Res 3, 23-30 (2022).
- M. Regardt et al., Hand exercise intervention in patients with polymyositis and dermatomyositis: a pilot study. Musculoskeletal Care 12, 160-172 (2014).
- M. A. Mattar et al., Exercise as an adjuvant treatment in persistent active polymyositis. J Clin Rheumatol 20, 11-15 (2014).
- A. N. Jorgensen et al., Effects of blood-flow restricted resistance training on mechanical muscle function and thigh lean mass in sIBM patients. Scand J Med Sci Sports 32, 359-371 (2022).
- A. Wallace et al., Community exercise is feasible for neuromuscular diseases and can improve aerobic capacity. Neurology 92, e1773-e1785 (2019).
- E. D. Johnson L, Walters S, Thickbroom G, Mastaglia FL. , Effectiveness of an Individualized, Home-Based Functional Exercise Program for Patients With Sporadic Inclusion Body Myositis. Journal of Clinical Neuromuscular Disease 8, 187-194 (2007).
- L. G. Johnson et al., Improvement in aerobic capacity after an exercise program in sporadic inclusion body myositis. J Clin Neuromuscul Dis 10, 178-184 (2009).
- E. Cerqueira, D. A. Marinho, H. P. Neiva, O. Lourenco, Inflammatory Effects of High and Moderate Intensity Exercise-A Systematic Review. Front Physiol 10, 1550 (2019).
- H. Alexanderson, C. Bostrom, Exercise therapy in patients with idiopathic inflammatory myopathies and systemic lupus erythematosus – A systematic literature review. Best Pract Res Clin Rheumatol 34, 101547 (2020).